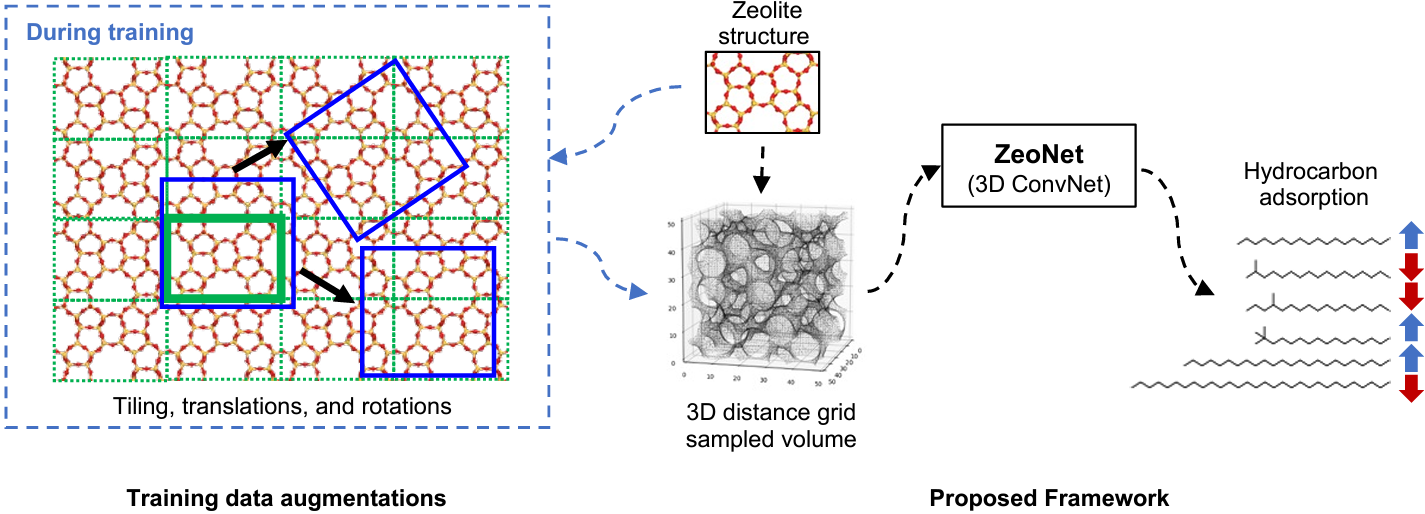
ZeoNet: 3D convolutional neural networks for predicting adsorption in nanoporous zeolites
Yachan Liu*, Gustavo Perez*, Zezhou Cheng, Aaron Sun, Samuel Hoover, Wei Fan, Subhransu Maji, Peng Bai (*equal contribution)
Github code repository
Zeolites are one of the most widely used materials in the chemical industry due to their nanometer-sized pores that can adsorb and react upon molecules selectively. With hundreds of known framework topologies and hundreds of thousands of computationally predicted structures, the ability to rapidly predict zeolite performance allows researchers to prioritize their efforts on the most promising structures for a given application. Although the accuracy of forcefield-based atomistic simulations has advanced significantly in the past two decades, these simulations can be computationally expensive, especially for long-chain, complex molecules. We present ZeoNet, a representation learning framework using convolutional neural networks (ConvNets) and 3D volumetric representations for predicting adsorption in zeolites. ZeoNet was trained on the task of predicting Henry’s constants for adsorption, kH, of n-octadecane in more than 330,000 known and predicted zeolite materials. Employing a 3D grid based on the distances to solvent-accessible surfaces, a volumetric representation that can be generated efficiently, the best-performing ZeoNet achieved a correlation coefficient r2 = 0.977 and a mean-squared error MSE=3.8 in ln kH, which corresponds to an error of 9.3 kJ/mol in adsorption free energy. In comparison, a model based on hand-designed geometric features has values of r2 = 0.777 and MSE=36.6. ZeoNet is also relatively efficient and can process ≈ 8 structures per second on an Nvidia RTX 2080TI GPU, orders of magnitude faster than forcefield-based simulations. A systematic analysis was conducted to investigate how the choice of ConvNet architectures, the linear dimension (L) and spatial resolution (∆d) of the distance grids, batch size, optimizer, and learning rate impact the model performance. We found that ConvNets based on the ResNet architecture offer the best tradeoff between expressiveness and efficiency. The performance for all models reaches a plateau at L = 30−45 A and depends less sensitively on grid resolution, with a small benefit around ∆d = 0.30−0.45 ̊A. Finally, saliency maps were visualized to identify which regions of the materials contributed the most to model predictions. It was found, interestingly, that the predictions are driven primarily by the accessible pore volume rather than the region occupied by the framework atoms.
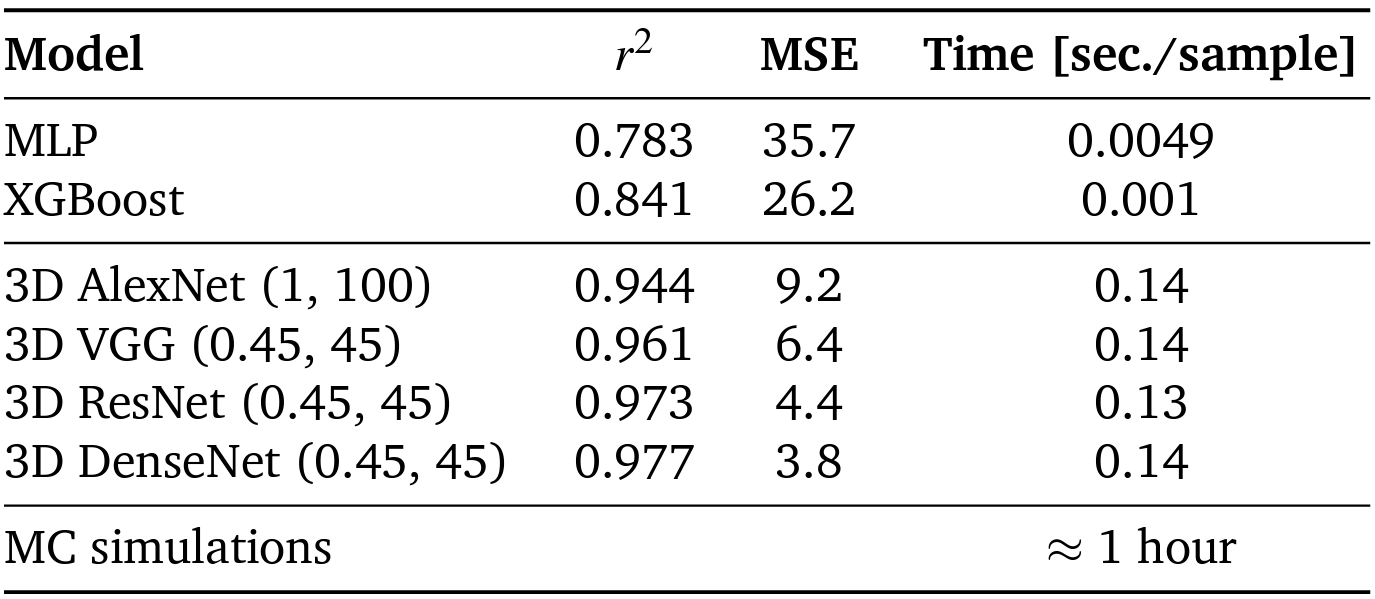
Tab. 1 Model performance for predicting C18 adsorption in zeolites, comparing the accuracy and efficiency of a multi-layer perceptron (MLP) and extreme Gradient boosting (XGBoost) trained on geometric features with various ZeoNet architectures operating on distance grids. The optimal input representations for each model are given in parentheses, (∆d,L) in Å.